RNAi-by-Soaking protocol
Ikuma Maeda and Asako Sugimoto
Updated on June 12, 2001
I-1. Preparation of DNA templates (For a small number of samples)
PCR reaction:
| yk cDNA phage lysate | 6 |
| 10 X PCR buffer | 10 |
T3 primer (A) (25pmol / 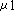 ) ) | 4 |
| T7 primer (A) (25pmol / ) | 4 |
| dNTPs (2.5mM each) | 8 |
| Taq polymerase | 0.5 (2.5U) |
| ddH2O | to 100 |
T3 primer (A): 5'-AATTAACCCTCACTAAAGGGAA-3'
T7 primer (A): 5'-GTAATACGACTCACTATAGGGC-3'
PCR condition:
| 94oC | 1' | |
| | 94oC | 1' |
2'30'' for 30 ~ 35 cycles |
| 55oC | 30'' |
| 72oC | 2'30'' |
| 72oC | 10' |
The PCR products can be used without any purification for in vitro transcription.
I-2. Preparation of DNA templates (For High-throughput RNAi)
Yuji Kohara's non-redundant cDNA set is sent as PCR product in 96-well format.
Each well contains ~ 1 of cDNA fragment solution.
Add 19 of H20 to each well.
We usually process 48 samples at a time, using 6 strips of 8 X 0.2ml tubes.
PCR reaction mix for 48 samples:
| (X50) | (per sample) |
| 10 X PCR buffer | 100 | 2 |
| T3 primer (B) (25pmol / ) | 40 | 0.8 |
| T7 primer (B) (25pmol / ) | 40 | 0.8 |
| dNTPs (25mM each) | 8 | 0.16 |
| Taq polymerase | 5 (25U) | 0.1 |
| ddH2O (RNase Free) | 767 | 15.34 |
| -------------------- | ----- | ------ |
| total | 960 | 19.2 |
T3 primer (B): 5'-AATTAACCCTCACTAAAGGGAACAAAAGCTGG-3'
T7 primer (B): 5'-GTAATACGACTCACTATAGGGCGAATTGGGTA-3'
Dispense 19.2 of the PCR reaction mix. Add 0.8 of the 1/20 diluted template DNA to each well.
PCR condition:
| 94oC | 5' | |
| | 94oC | 1' | for 35 cycles |
| 64oC | 30'' |
| 72oC | 2'30'' |
| 72oC | 10' | |
Use the PCR product directly to in vitro transcription.
II-1. RNA synthesis (For a small number of samples)
in vitro transcription:
| 5 X transcription buffer | 20 |
| 1M DTT | 3 |
| NTPs (2.5mM each) | 20 |
| PCR product (from step I-1) | 30 |
| T7 or T3 polymerase | 40U (STRATAGENE) |
| ddH2O | up to 100 |
- Mix well and incubate at 37oC for 2 to 3 hrs.
(RNA products can be checked by running ~ 1 on denaturing agarose gel at this point.)
- Mix the sense and antisense RNAs in the same tube. Total volume becomes to 200. DNase treatment is not necessary.
- Extract with phenol, phenol/chloroform, and chloroform.
- Add 0.1 volume of 3M sodium acetate pH 5.2 and 0.6 volume of isopropanol to precipitate RNA. Spin mixtures at room temperature.
- Rinse the pellet with 70% EtOH.
- Dry up and resuspend the pellet in 16 of the Soaking buffer. The RNA solution may become somewhat viscous, but it will not be harmful.
- Check the quality of the RNA solution by running 0.5 on denaturing agarose gel. Final RNA concentration will be 1 ~ 5
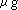 / . Annealing is not necessary.
/ . Annealing is not necessary.
Soaking buffer:
0.25X M9 (Mg++ free)*
3mM spermidine (SIGMA S2626)
0.05% gelatine (autoclaved and filtered)**
**if you use siliconized tubes, this can be omitted.
* 5X M9(Mg++ free)
| Na2HPO4 | 3.4g |
| KH2PO4 | 1.5g |
| NaCl | 0.25g |
| NH4Cl | 0.5g / 100ml |
II-2. RNA synthesis (For High-throughput RNAi)
in vitro transcription for 48 samples:
| (X50) | (per sample) |
| 5 X transcription buffer | 250 | 5 |
| 1M DTT | 37.5 | 0.75 |
| NTPs (25mM each) | 25 | 0.5 |
| T7 or T3 polymerase (50U/ul) | 10 | 0.2 |
| ddH2O | 552.5 | 11.05 |
| -------------------- | ----- | ------ |
| total | 875 | 17.5 |
- Dispense 17.5 of the transcription mix into a 6 strips of 8 X 0.2ml tubes, and add 7.5 of the template DNA (=PCR reaction mix from step I-2).
- Mix well and incubate at 37oC for 2-3 hours.
(Check the RNA product by running ~1 on a denaturing agarose gel.)
- Combine the sense and antisense RNA solutions, using an 8-channel pipette. Total volume will be 50.
- Add 50 of TE-saturated phenol to each tube, and vortex for 10 min.
- Centrifuge (3,000 rpm, or maximum speed) with a swing rotor for microtiter plates at room temperature for 30 min.
- Using an 8-channel pipette, remove the BOTTOM (organic) phase.
- Add 50 of phenol-chloroform to each tube, vortex, and centrifuge as above.
- Using an 8-channel pipette, remove the BOTTOM (organic) phase.
- Add 50 of chloroform to each tube, vortex, and centrifuge as above.
- Carefully transfer the TOP (aqueous) phase to a new 8 X 0.2ml strip, using a single-channel pipette. (It might help to do it under a dissecting microscope!)
- Add 4 of 3M sodium acetate (pH 5.2) and 26.5 of isopropanol and vortex.
- Centrifuge at 25oC for 35 min. at a maximum speed (3000rpm for our swing-rotor for microtiter plate).
- Rinse with 200 of 70% EtOH.
- Centrifuge as above. Remove supernatant and dry up.
- Resuspend with 4 of 1X soaking buffer.
- Check the integrity of RNA by running 0.5 of the sample on an agarose gel.
- Anneal the dsRNA by heating 68oC for 10 min., then 37oC at 30min. (You may omit this step.)
- Store the dsRNA at -80oC
III-1. Soaking (For analyses of F1 progeny and P0 germline)
- Wash L4-stage hermaphrodites well in M9.
- Transfer the worms to a fresh NGM plate without bacteria, and let them crawl for several minutes to remove bacteria completely.
- Transfer 4 of RNA solution into a 200 PCR tube.
- Pick four~eight worms and put them into the RNA solution, then incubate at 20oC for 24 hrs.
- Recover them onto new NGM plates with bacteria. Transfer the P0s to new plates every 24 hrs. (We rear the worms at 25oC so that temperature sensitive phenotypes can be detected.)
- Observe the phenotypes of F1 progeny and P0 worms.
(In many cases, first F1 animals laid just after recovery are fully affected with RNAi.)
III-2. Soaking (For analyses of postembryonic phenotypes)
You can examine the postembryonic phenotype by soaking L1 larvae in the dsRNA solutions.
- Collect 10-20 gravid hermaphrodites and wash them with M9 (contains 0.05% gelatin).
- Cut them open and collect embryos.
- Transfer embryos in M9 to a 1.5ml tube then incubate at 25oC overnight.
- Transfer hatched L1 worms to a fresh NGM plate without bacteria and let them crawl for several minutes to clean them up.
- Pick 15-20 worms and soak them into RNA solution (4 in a 200 PCR tube), then incubate at 20oC for 24 hrs.
- Recover them to new bacterial plates.